Transition metal hydrides are ubiquitous intermediates in catalytic systems and the transfer of a hydride from the metal center to an organic substrate is the turnover limiting step in many catalytic processes. For example, the transfer to a metal hydride to CO2 is important in many reactions that utilize CO2 as a feedstock. Given its relevance to a variety of transition metal mediated process, we seek to gain fundamental understanding about the factors that control hydride transfer from a transition metal to an organic substrate. We have recently reported a comprehensive linear free energy relationship (LFER) between two intrinsic parameters which govern the propensity of a transition metal hydride to donate a hydride ion: thermodynamic and kinetic hydricity. Thermodynamic hydricity (Figure 1a) is the required free energy to release a free hydride ion, H-, from a species in solution (ΔG°H-), and kinetic hydricity (Figure 1b) is the rate constant for a hydride transfer reaction and is related to the free energy of activation (ΔG‡H-). The development of this LFER presents a unique opportunity to study effects that may break the scaling relationship between kinetic and thermodynamic hydricity. Of particular interest is understanding the potential influence of functional groups in the secondary coordination sphere that may either i) participate in hydrogen bonding, or, ii) exert an electric field gradient effect. Further, we hypothesize that hydride transfer reactions involving transition metal hydride donors can be modelled using Marcus theory. We are currently investigating this proposal using kinetic and computational studies and expect that the insight provided by this work will enable us to design improved catalysts for hydride transfer reactions.
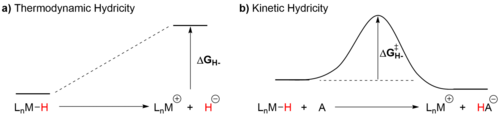
Figure 1. Representations of: a) Thermodynamic hydricity, which is the Gibbs free energy difference between the ground state of the metal hydride (LnM-H) and the product of heterolytic bond scission (LnM+ and H‑). b) Kinetic hydricity, which is the elementary rate constant for hydride transfer from a metal hydride (LnM-H) to a hydride acceptor (A).