Concerns about the environmental consequences of continued fossil fuel use for the synthesis of commodity chemicals has led to a search for alternative carbon sources which are sustainable. CO2 is an attractive feedstock due to its high abundance, low cost and toxicity, and relative ease of transport. However, the catalytic conversion of CO2 is complicated by its high kinetic and thermodynamic stabilities. A potential solution to this problem is to utilize transition metal complexes, which can interact with CO2 and weaken the strong C=O double bonds. This can provide a low energy pathway for the conversion of CO2 into value-added products. Nevertheless, at this stage the number of chemicals that can be produced using CO2 as a feedstock is small. In collaboration with Professor Wesley Bernskoetter at the University of Missouri, we have developed state-of-the-art iron catalysts for the hydrogenation of CO2 to formate (Figure 1) and methanol.
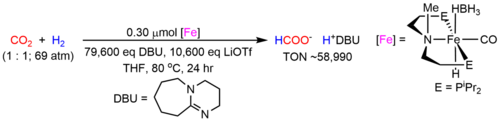
Figure 1. State-of-the art iron catalysts for the hydrogenation of CO2 to formate. A related catalyst is active for the hydrogenation of CO2 to methanol.
Currently, we are focusing on improving the performance of our catalysts for CO2 conversion to methanol and also developing systems that can convert CO2 and other basic feedstocks such as olefins and alcohols into carboxylic acid esters.